


URGENT : SÉCURITÉ SUR LE TERRAIN

Dispositif : Système d’endoprothèse thoracique Relay Pro dans la configuration de l’endoprothèse couverte (Non-Bare Stent) >= diamètre
32 mm (dispositifs 28-N4)
À nos estimés clients,
Cette lettre a pour but d’informer les dépositaires que Bolton Medical Inc. (filiale de Terumo Aortic) procède à un rappel de dispositif médical volontaire (correction) relatif à des changements de fabrication et une mise à jour des étiquettes pour tous les lots du système d’endoprothèse thoracique Relay Pro, N4 : configuration de l’endoprothèse couverte 32 mm et diamètres supérieurs. Des cas d’échec de relâchement de l’endoprothèse partiellement déployée se sont produits. Veuillez noter que ce mode de défaillance peut se produire sans signe préalable et qu’aucune méthode de sauvetage fondée sur le dispositif n’a été identifiée pour ce scénario particulier. Nous vous conseillons d’envisager d’autres options d’endoprothèse avant d’utiliser les dispositifs Relay Pro concernés, jusqu’à ce que l’évaluation de la cause fondamentale soit terminée et que des mesures d’atténuation efficaces soient en place. Dans les cas où le Relay Pro est utilisé, si ce problème se pose, le jugement clinique doit guider la prise de décision en temps opportun, y compris en envisageant la conversion en chirurgie ouverte, le cas échéant.
Description du problème :
Dans le cadre de la surveillance après la commercialisation, nous avons reçu des plaintes selon lesquelles la prothèse ne peut pas se détacher du système de déploiement parce que la pince proximale est déconnectée du tube de contrôle externe. L’utilisateur peut le remarquer du fait d’un manque de résistance ressenti lors du glissement arrière du support d’apex, accompagné d’un défaut de relâchement de l’endoprothèse proximale. L’implant ne peut pas être récupéré à ce stade de la procédure.
Notre enquête préliminaire nous a permis de déterminer que la conception et la fabrication pourraient contribuer à ce problème du dispositif. L’enquête sur la cause fondamentale étant en cours, les dispositifs/modèles concernés et les mesures correctives peuvent changer.
Risque pour la santé :
Bolton Medical a reçu quatre (4) plaintes au cours des huit (8) derniers mois associées à ce mode de défaillance, dont trois (3) ont entraîné la mort, y compris (1) perforation de l’aorte, et deux (2) conversions en chirurgie ouverte qui ont entraîné la mort du patient en raison d’un accident vasculaire cérébral. Dans ces cas, les pinces proximales sont déconnectées du tube de tube de contrôle externe. Cela a empêché la libération des endoprothèses par le système de déploiement.
Ce mode de défaillance ne peut pas être reconnu avant sa survenue au cours de la procédure. Les difficultés à libérer l’endoprothèse peuvent entraîner un retard de la procédure, un déplacement de l’endoprothèse et l’incapacité de libérer l’endoprothèse. Cela peut nécessiter une conversion en chirurgie de répartition ouverte afin de libérer la pince et peut entraîner la mort du patient.
Mesure corrective :
En cas de difficulté à libérer l’endoprothèse proximale et d’absence de résistance lors du glissement arrière du support d’apex, nous recommandons à l’utilisateur de commencer par essayer les techniques de sauvetage existantes décrites dans le mode d’emploi avant de s’engager dans une autre ligne de conduite. Si le problème persiste, aucune autre méthode de sauvetage du dispositif n’est disponible et le jugement clinique devrait guider la prise de décision en temps opportun, y compris en envisageant la conversion en chirurgie ouverte. Les mises à jour des étiquettes seront communiquées à mesure que d’autres renseignements seront disponibles.
Les mesures correctives immédiates prises par Terumo Aortic comprennent des changements de fabrication visant à réduire la probabilité de défaillance de l’attache du système de déploiement. Les dispositifs de la famille Relay Pro N4 qui intègrent des configurations de fils-guides (tailles supérieures ou égales à 32 mm de diamètre) ont été identifiés comme présentant un risque accru concernant le mode de défaillance signalé pour les raisons suivantes :
- L’enquête a permis de déterminer que l’élément principal du processus de fabrication menant au mode de défaillance n’est utilisé que pour le N4 en configuration d’endoprothèse couverte avec fils-guides >= 32 mm.
- Les plaintes confirmées sont limitées aux dispositifs RelayPro N4 avec fils-guides.
- Aucune défaillance de ce type confirmée sur terrain n’a été identifiée dans les autres familles de produits.
À l’heure actuelle, il y a peu d’information pour appuyer l’efficacité de ces mesures correctives, étant donné que l’enquête sur la cause fondamentale est toujours en cours. Les actions correctives futures peuvent inclure d’autres modifications de fabrication et de conception.
Appareils inclus :
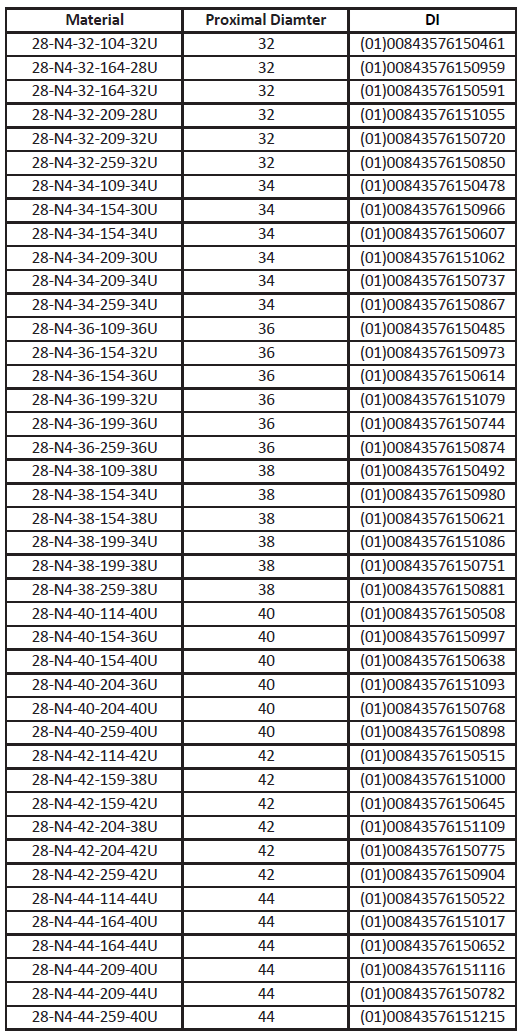
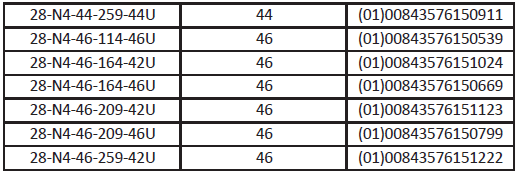
Mesures à prendre par le client :
- Envisager d’autres options d’endoprothèse avant d’utiliser les dispositifs Relay Pro concernés, jusqu’à ce que l’évaluation de la cause fondamentale soit terminée et que des mesures d’atténuation efficaces soient en place.
- Informer tous les utilisateurs de Relay Pro des conseils supplémentaires pour la gestion des cas où l’endoprothèse ne peut être libérée du système de déploiement.
- Veuillez afficher une copie de cet avis à l’endroit où les appareils sont stockés et la conserver avec le mode d’emploi.
- Remplir l’annexe 1 accusant réception et communication de cet avis de correction de dispositif médical. Retourner l’annexe 1 à Terumo6732@sedgwick.com.
- Si des appareils ont été transférés à un autre établissement, veuillez leur fournir une copie de la notification et leur demander de suivre les mesures décrites dans la présente section.
Soyez assuré que nous prenons la sécurité et la qualité de nos produits très au sérieux et que nous restons engagés à vous soutenir par des conseils clairs et un suivi continu. Si vous avez d’autres questions, veuillez communiquer avec votre représentant de Terumo Aortic ou écrire à us Market_Actions-TMC@terumomedical.com.
Les événements indésirables peuvent être signalés au programme de déclaration d’événements indésirables MedWatch de la FDA par :
Internet : site Web de MedWatch à l’adresse http://www.fda.gov/medwatch
Téléphone : 1-800-FDA-1088 (1-800-332-1088)
Courrier : MedWatch, HF-2, FDA, 5600 Fishermen’s Lane, Rockville, MD 20852-9787
Au nom de Bolton Medical, Inc.
Kimberly Feitl
Vice-présidente, Qualité
URGENT : SÉCURITÉ SUR LE TERRAIN
Annexe 1 : Formulaire d’accusé de réception d’un avis de correction d’un dispositif médical
FSN2026-002
Renseignements sur le compte :
Nom du client : |
|
Adresse du client : |
|
Numéro de téléphone du client : |
|
Adresse courriel du client : |
|
En signant ci-dessous, je reconnais que :
· J’ai reçu l’avis de correction d’un dispositif médical et je confirme que j’en comprends le contenu.
J’ai communiqué l’information aux utilisateurs de mon organisation.
Nom (en lettres moulées) : |
|
Signature : |
|
Date : |
|
Veuillez retourner le formulaire dûment rempli à Terumo6732@sedgwick.com.